The FDA is the GMP enforcement agency in the US, wielding both the FDA Form 483 and Warning Letters to ensure compliance.

Understanding and Responding to an FDA 483
Form 483s are “inspectional observations” conducted by FDA auditors that serve the purpose of documenting and communicating any discovered manufacturing concerns. Although a formal response is not mandated, a Form 483 should be promptly and thoroughly addressed. Pharmaceutical companies that receive a Form 483 should attempt to gather as much information as possible including:
- Understanding the observations and validating their accuracy
- Understanding the “broader message” the FDA is sending
- Identifying awareness of applicable regulations
- Consulting with legal counsel as necessary.
Along with its observations, the FDA will also consider an Establishment Inspection Report as it considers any further action it may need to take. After receiving an FDA 483, companies should work closely with the FDA in order to remedy all identified issues.
After a Form 483 is issued and the inspector completes the Establishment Inspection Report, the agency may issue a Warning Letter as a subsequent measure. A Warning Letter indicates that higher FDA officials have reviewed the observations, confirming that a serious violation may exist. While this is considered a worst-case scenario, the FDA has repeatedly indicated that it will not hesitate to use a Warning Letter if compelled to do so. In the last fiscal year alone (October 2018 to September 2019), 81 Warning Letters were issued by the FDA.
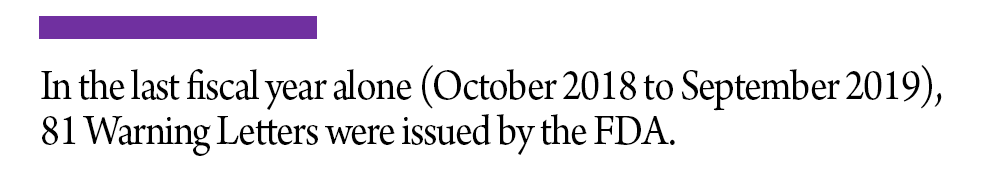
In fact, there may even be indications that this is a growing trend, as this is the highest number over a five-year period (the Warning Letters addressed to Compounders or Compounding Pharmacies are not included in this analysis).
A Warning Letter should be avoided at all costs. Thus, an FDA observation should be treated as a prime opportunity to correct non-compliance, designated as a high-level priority from the C-suite to the manufacturing floor. A Warning Letter puts a pharmaceutical in a vulnerable position with a range of consequences that can include:
- Safety: Delivering unsafe medication that may cause unwanted side-effects, injury, or even death
- Public relations fallout: The public, shareholders, and other stakeholders can react negatively, potentially affecting stock prices and consumer confidence
- Loss of marketing authorization: The drug can be pulled from the market, representing potentially millions in lost sales
- Response(s) from competitors: A competing pharmaceutical can take advantage of negative publicity, using it to create a market opportunity for themselves
- Future impact on new drug approvals: The FDA can put a hold on any new drug approvals
If an FDA 483 is not promptly managed, its observations can be quickly escalated via a Warning Letter, resulting in subsequent negative consequences which could be detrimental to the drug’s successful future.

Avoiding a Warning Letter
From the inception of the FDA more than 100 years ago, manufacturing operations have been subject to a near-constant evolution of technologies, processes, and regulations. This perpetual advancement also comes with a hefty price tag which, in turn, makes it more challenging to protect investments.
When FDA 483 observations are handed down, ROI is threatened as normal operations are significantly disrupted. All staff previously devoted to the manufacturing cycle should now be involved in the response in order to mitigate the risk of a Warning Letter. This collective approach also assists in resuming normal operations as soon as possible.
With everyone involved in an FDA observation and response, costs can quickly mount with various companies citing ranges from $250,000 to tens of millions of dollars in lost production (depending on the scope of the observation). To best navigate an FDA 483, mitigate consumer harm, and best protect ROI, pharmaceutical companies should pursue the following actions:
- Work with a knowledgeable partner
Receiving a 483 or Warning Letter requires a thorough, well-researched, and well-planned response that embraces transparency. At this point, it is advisable to work with a deeply knowledgeable partner. Firms would be well-served to cooperate with partners which specialize in the policies developed by regulatory agencies while also maintaining a focus on the company’s specific operations. - Implement SOPs and regular QA inspections and reviews
Clear, detailed, and visible guidelines related to all operations are not only essential for GMP but also for applicable regulatory requirements as a whole. Standard Operating Procedures (SOPs), which are critical in ensuring adherence to GMP, are within the FDA’s scope and should be on any pharmaceutical manufacturer’s radar. Investing in the development of SOPs enable better consistency in operations.

While there are many examples available of thorough SOPs, best practices are those developed collaborating with key stakeholders for both manufacturing and QA/QC. SOPs, with clear QA/QC controls, can potentially mitigate both mechanical as well as human and handling errors that can prompt observations and other events. - Collaborate with the FDA
It is important to remember that the FDA shares the same goals as the pharmaceutical industry and can provide a tremendous amount of assistance in resolving a 483. Response to a 483 is just one aspect of a pharmaceutical company’s relationship with the FDA. It is wise to communicate frequently and transparently in order to maintain a positive and constructive relationship. - Proactively avoid the root causes of an FDA 483
The simplest method of avoiding a Warning Letter is taking proactive steps to avoid an FDA 483. Rather than responding to the potentially unsettling results of an inspection, it is better to preemptively root out all possible sources of non-compliance. The surest way to achieve this would be to work with third party suppliers or “compliance services” providers which are uniquely suited to minimizing any missteps which could bring about an FDA 483. Such entities provide a range of services, the foremost of which are mock audits meant to best simulate FDA inspections.

The mock inspections are intended to uncover all sources of non-compliance that might later be noted by FDA auditors, allowing firms the opportunity to fix the problem before it even arises. The specialists or service providers, besides discovering any issue(s) also typically provide a range of next steps to mitigate the problem(s) and thus avoid unfavorable consequences.
It is also prudent to stay up-to-date on the evolving trends of the industry and associated regulatory policies and guidelines to avoid non-compliance due to ignorance. Also, with evolving technological advancements effecting efficiency, reliability, productivity, and compliance of pharmaceutical manufacturing, staying up to date and implementing such practices becomes essential. Simultaneously, it is crucial to ensure that employees are trained on the use of modern instrumentation and emerging technologies to stay at par with changing industry standards.
Conclusion
An FDA Warning Letter should be avoided at all costs to avoid potential consumer harm, negative public response, and the financial fall-out that would likely result from publicly communicated non-compliance.
While it goes without saying that an FDA 483 and Warning Letter should both be avoided, the former can be expertly managed, saving time, money, and, ultimately, preserving public health and safety.
Through the implementation of detailed SOPs within manufacturing workflows and its integration with QA/QC, as well as working with a knowledgeable partner, can ensure best practices that are compliant and follow the regulatory guidelines to avoid an observation by the regulatory agency.
REFERENCES
- U.S. Food and Drug Administration. FDA Form 483 Frequently Asked Questions as referenced at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/fda-form-483-frequently-asked-questions
- Wikipedia. FDA Warning Letter as referenced at https://en.wikipedia.org/wiki/FDA_warning_letter
- Panko, Ben. Where Did the FDA Come From, And What Does It Do? Smithsonian.com. February 8, 2017 as referenced at: https://www.smithsonianmag.com/science-nature/origins-FDA-what-does-it-do-180962054/
- Unger, Barbara. An Analysis of 2017 FDA Warning Letters on Data Integrity. Pharmaceutical Online. May 18, 2018 as referenced at https://www.pharmaceuticalonline.com/doc/an-analysis-of-fda-warning-letters-on-data-integrity-0003
- U.S. Food & Drug Administration. CFR – Code of Federal Regulations Title 21 as referenced at https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=11
- Jones Day. Current Trends in the FDA’s Enforcement of GMP Requirements. March 2013 as referenced at https://www.jonesday.com/en/insights/2013/03/current-trends-in-the-fdas-enforcement-of-gmp-requirements
- Food and Drug Administration. When and why was the FDA formed? March 2018 as referenced at https://www.fda.gov/about-fda/fda-basics/when-and-why-was-fda-formed
- Otto, Ralf; Santagostino, Alberto; and, Schrader, Ulf. Rapid growth in biopharma: Challenges and opportunities. McKinsey & Company. December 2014 as referenced at https://www.mckinsey.com/industries/pharmaceuticals-and-medical-products/our-insights/rapid-growth-in-biopharma
- U.S. Food & Drug Administration. Data Integrity and Compliance With Drug CGMP Questions and Answers Guidance for Industry; Page 4. December 2018 as referenced at https://www.fda.gov/media/119267/download
- Chen, Tony. A Bad 483 Could Cost a Company Millions. FDAzilla. August, 18, 2016 as referenced at https://blog.fdazilla.com/2018/08/what-does-getting-an-483-or-warning-letter-really-cost-you/
- Rothrauff, Drexel. Top 5 Immeasurable Costs of a Warning Letter. Blue Mountain Quality Resources. October 14, 2013 as referenced at https://www.coolblue.com/blog/industry-insights/top-5-immeasurable-costs-of-a-warning-letter/
- Miller, Bob. FDA Form 483. Healthcare Consultants Pharmacy Staffing. May 27, 2019 as referenced at https://pharmacy-staffing.com/fda-form-483/
- Land, Mark. The Importance of Standard Operating Procedures. American Association of Homeopathic Pharmacists. November 9, 2016 as referenced at https://www.theaahp.org/compliance/the-importance-of-standard-operating-procedures/
- European Medicines Agency. Good manufacturing practice. As referenced at https://www.ema.europa.eu/en/human-regulatory/research-development/compliance/good-manufacturing-practice
- ECA Academy. FDA Inspection Reports: What is What. July 8, 2015 as referenced at https://www.gmp-compliance.org/gmp-news/fda-inspection-reports-what-is-what
- Imarc. FDA Form 483 and Warning Letters: What’s the Difference? April 30, 2019 as referenced at https://www.imarcresearch.com/blog/form-483-and-warning-letters
- U.S.Food and Drug Administration. Warning Letters. November 26, 2019 as referenced at https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters
- FDAZILLA. The Ultimate Guide To Form FDA 483s. February 5, 2019 as referenced athttps://blog.fdazilla.com/2019/02/fsma-pharma-medical-devices-the-ultimate-guide-to-form-fda-483s/
